- 近期网站停站换新具体说明
- 按以上说明时间,延期一周至网站时间26-27左右。具体实施前两天会在此提前通知具体实施时间
主题:【原创】中日欧非印人类全基因组数据的聚类图 -- 瓦斯
这是一个纯属个人业余好奇所做的事情。
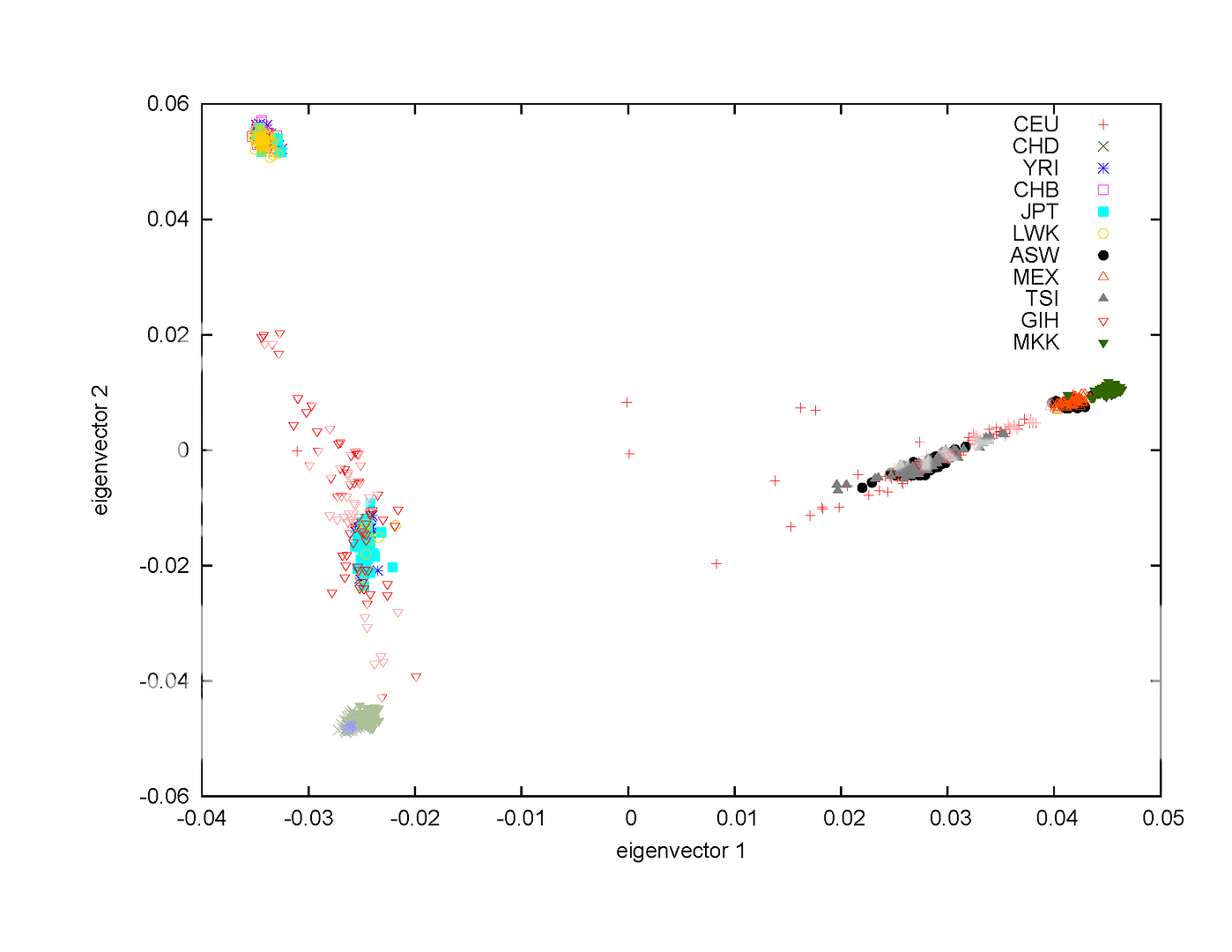
原始数据下载自[URL=ftp://ftp.ncbi.nlm.nih.gov/hapmap/raw_data/hapmap3_affy6.0/]ftp://ftp.ncbi.nlm.nih.gov/hapmap/raw_data/hapmap3_affy6.0/[/URL]。压缩文件总计大小约37G。包含中日欧非印等11个人类群体,总人数约1300人。
解压后可以得到每个人的二进制原始数据,每个文件约66M,每个人都有全基因组一百万个位点的基因型数据,总计大小约81G。
这是11个群体的缩写:
ASW African ancestry in Southwest USA
CEU Utah residents with Northern and Western European ancestry from the CEPH collection
CHB Han Chinese in Beijing, China
CHD Chinese in Metropolitan Denver, Colorado
GIH Gujarati Indians in Houston, Texas
JPT Japanese in Tokyo, Japan
LWK Luhya in Webuye, Kenya
MEX Mexican ancestry in Los Angeles, California
MKK Maasai in Kinyawa, Kenya
TSI Toscans in Italy
YRI Yoruba in Ibadan, Nigeria
本图采用Nature 456, 98-101 (6 November 2008)文中描述的软件制作。
本帖一共被 1 帖 引用 (帖内工具实现)
这张图是根据一个统计方法的计算结果画出来的。这个统计方法叫主成分分析方法(Principal Component Analysis,简称PCA)。外链出处
分析的数据是11个人类群体,总人数约1300人的基因突变数据,也就是在人类基因组上一百万个常见有突变的位点通过实验分析得出的基因型数据。每个人的每个位点上有两个等位基因:一个从父亲来,另一个从母亲来。所以基因型数据只有三种可能:要么不存在突变,两个等位基因都正常;要么有一个突变另一个正常;要么两个都是突变,可用代码0、1、2分别代表。这样每个人在一百万个位点上就有一百万个基因型代码数据,1300人就有十三亿个数据。
使用主成分分析方法分析不同人类群体,目的是看看不同人类群体的基因差距有多大,有哪些突变决定了群体差异。分析结果可用于人类进化分析,也用于辅助寻找疾病相关基因。当然,相信基因大战或基因武器的,也可以梦想这个结果可用于此类目的。
主成分分析方法很复杂,我也不懂,只能说点儿皮毛。先从一个位点的突变看,数据有11个人类群体,大致平均每个群体有100人左右,根据每个群体中每个人在这个位点上基因型可以计算出突变在该群体的发生频率。不同的人类群体,突变频率可能有差别。也可能差别不大,不太可能明确判断。很自然会想到用两个位点的突变判断比一个位点好,这样一来数据就成了二维的;用三个位点判断,就是三维的分析。。。一百万个位点当然就是一百万维的分析,只不过这时早晕菜了。另外,并不是一百万个位点的每个突变在人类群体中有差别,都可以用来分析群体。就是说,那一百万维不是都有用,分析时应尽量用有用的来分析。这也就是通常说的降维。
主成分分析方法的实质就是降维,将人类群体基因突变差异经过转化,将差异最大程度地集中于较少的空间向量(维)。这张图就是用两个集中了最大突变差异的空间向量(eigenvector)画的。但这张图只是个相对粗略的分析结果,有不同的人类群体没有分开,但也有同一群体的样本却被分开了。
最后说明一下,我不是搞统计的,只能胡侃。有说错的地方,请及时纠正。
送花感谢。
标准操作,但结论真不好说
偶也不是搞统计的,但是用的软件里涉及一些这方面的内容,所以稍微知道,业余的白话一下。对于一个大型的高维数组,想要搞清楚他们的分类并且形象的表示出来很难,所以通过某种算法,将其按照相互间性质彼此靠近程度来转换到一个低维的数组来。在这里,我觉得每一个人的基因的位点就是原始数据维数,最后变换为一个2维的数组显示在上述的图表里。可以理解为,将高维数组按照内部性质彼此靠近程度来投影到平面上,这样在平面上越靠近的点(就是人,一个人一个点)就说明对应的原始数据(基因)越靠近
这些群体间达到了多少百分比(横坐标和纵坐标百分值之和)的差异。应该还有一张图对应说明是哪些成分或者成分的组合解释了这些群体间的差异。
我也不太懂,说的多了就露短了。但仅一个图给出的信息是不完整的。
我是比较懒,所以给了图,大致说说就算了,只有碰到懂门道的才切磋一下。前5个主分量分别是:
65.7740
31.9590
7.5320
6.8140
6.1620
是啊,是要有一个说明第一和第二主分量分别代表了哪些位点的。不过都是1百万个位点中的一部分,列出来也没有什么意思,就懒得弄了。最终还是图示一目了然。
应先过滤一下,尽可能将没用的位点(noninformative)排除掉再分析。如先计算每个位点在不同群体频率的平均值和CV,再根据CV的分布决定过滤的标准。
现在的软件里面都带有默认的过滤筛选过程,自己要亲手搞搞修改一下也是可以的。
这么直接用全部SNP数据,没有统计校正么?
统计校正在那个软件都有自动的过程。
做这个事情,主要是费时在:要把数据按照软件要求的格式准备好。如果都准备好了,实际计算时间就是100小时左右。可是我花了有半个月时间吧,断断续续的,经常是运行了一两天才跳出来说哪个地方不对,你又要从头开始修改准备格式,这样折腾了很多次才得到结果。
在开放读码框(ORF)里的,在内含子里的,在基因侧翼区的,等等。。。更别说同在ORF里,密码子第一二位和第三位也有所不同。
不同的位点承受的选择压力不同时,不好放在一起比的。因为hapmap不是把人基因组中所有的SNP都找全了,所以他的SNP数据本身就有偏异的。这个东西叫Ascertainment Bias。是必须被校正的。
我倒是很佩服您的耐心。花这么长时间做这个图。非有大兴趣大耐心者不能为。我若能献花,一定献一个以表敬意。
日本人群在第二主成份上分成明显的两群。一个和中国人接近,一个和德州印第安人接近。这个说明什么呢?虾夷人么。