- 近期网站停站换新具体说明
- 按以上说明时间,延期一周至网站时间26-27左右。具体实施前两天会在此提前通知具体实施时间
主题:【原创】什么是RCT--- 送给中间派的您 -- 虽远必诛
这个不是问题,可以做成肠溶胶囊,这样不影响吸收,也不会被患者发现。
除非您认为,中药的有效是汤药中的热水。
汤药作为液体制剂,其吸收过程从胃部即已开始,制成肠溶制剂的理论依据何在?
用胶囊一类的固体制剂验证液体制剂的疗效,本身就不是等效性比较。说一句,改变剂型按规定也要做临床试验。
实际上,对于新药研发,处方都是固定的,根本不存在汤剂的问题。最接近汤剂的液体口服制剂是合剂。它的安慰剂不难做。俺工作后接的第一个活儿就是合剂安慰剂
汤药类似于西医的个体化给药方案,但处方不固定,更为复杂。请教一下,对于氨茶碱之类药物的个体化给药方案有没有做过大规模临床试验?试验方案是怎么设计的?如有可能,请做些介绍,窃以为对中药的临床方案设计研究更有助益。
剥夺江湖骗子的饭碗。
胃仅仅吸收少量的糖,酒精和水分,大多数的消化吸收是在小肠。
这外行还真是难缠。
现在没有的自然需要进一步试验,这也是为什么要大规模试验的原因。
西药特殊需要量化的多了去了,一样也可以。
请搞清楚基本常识先。
您对药物的体内过程缺乏最基本的了解。
首先,小肠是口服药物吸收的主要部位,但不等于药物只在小肠吸收。很多药物在胃即已开始吸收过程,差别只在吸收速率的不同。
其次,即使您研究的药物在胃内完全不吸收,但是,胃的排空速率,胃的蠕动,胃内食物的多少都会影响到药物的吸收过程,甚至某些遇酸遇酶不稳定的药物会在胃中降解。也就是说,普通口服制剂改为肠溶制剂将完全改变其体内吸收分布过程,吸收的快慢、多少甚至在体内停留的时间都不同,从而影响疗效。
那么,把汤剂改成普通的胶囊行不行呢?答案还是——不行。这是因为固体制剂必须在消化道内先崩解成小颗粒,再溶解,然后才进入吸收过程。聪明如您,必能看出,固体制剂吸收永远比液体制剂慢,也即——胶囊永远比汤剂吸收慢,吸收分布过程也不一样,对于药物评价,这种差异是不可接受的。
这就是为什么一种已经批准上市,确认安全有效的药物如果改变剂型,在中国必须要求重新做药理试验和临床试验的原因——估计虽远大夫待的国家也有同样要求。
多说一句,中药复方汤剂制成胶囊剂,很多时候是不可能完成的任务。即使可能做到,和原汤剂相比,肯定不具有等效性。医生缺乏药学知识,不知道情有可原。
把别人的话极端化没有意思,大家都清楚,吸收主要在肠,少量在胃。不要抬杠。
至于说不同剂型,药代也清楚的很,参加过药品的临床试验。
倒是比较感兴趣,你汤剂如何能够做到不让患者感到同安慰剂的区别?
而且还要保证安慰剂没有任何生理作用?
色,味,形完全一样,没有生理作用,确实比较高明。
真心请教。
很多试验的患者是相互看对方吃什么药的,这样安慰剂要求就非常严格了。
科普文章,首先要保证内容正确,不要灌输错误概念。
从您的回答看出,您对药代过程理解存在误区,对不同剂型的差别更不甚了了。
用固体制剂代替液体制剂,用肠溶制剂代替普通制剂,一上来就两个根本性错误。您的临床方案打根儿上就是错的,后面双盲随机设计再完美,观察指标再科学有啥用?
拿化药举个例子,要评价阿司匹林泡腾片的疗效和安全性,您拿阿司匹林肠溶片作为试验药物,能行吗?得出结论说阿司匹林肠溶片对胃粘膜无刺激性,所以泡腾片也对胃粘膜无刺激性,您觉得可笑不?
我要是纠缠于下胃管啊鼻饲啊才叫抬杠,谁都知道这是无实际操作性可言的,说说而已罢了。
安慰剂的问题,对干这行的觉得简单,但解释起来也不是一两句话能说清楚的。简单地说,制作时除了采用相同的辅料,适当的色素和矫味剂之外,还要加入一些无药偏性的原料,通常是食品类成分。加入什么原料,首先需要取得药理和临床专家认可,再从制剂角度考虑。制成的安慰剂也要和试验药分别编号,作各种各样的指标评价,涉及到人的主观评价的,也一样采用双盲法对照评价。对于中药,安慰剂和试验药物完全一致很难做到(有时候试验药物自身性状都不一致),但可以做到尽可能接近,象“中药”。
你也知道,中药的安慰剂无法达到不能分辨的地步。那样安慰剂效应就无法避免。
为什么中药研究的 methodology 都不好,从根本上讲无法回避安慰剂效应。
中药,压片包 薄膜衣的很多, 但是到了胃内还是会让患者感觉到,尤其是刺激性比较大的药物。这样患者就知道自己吃的是药物,还是安慰剂。
这样的临床有多大价值?非常低。
汤药就更无法避免了。
止于说不同剂型之间的差异,中药远没有西药明显。
止于说临床方案,不是你说了算,我说了算,是要拿出来大家peer review。
你说中药的RCT,必然要解释用什么做对照,如何避免安慰剂效应的问题。
止于说不同剂型,先把安慰剂效应搞清楚再说吧!
还是那句话,试验药物打一开始就搞错了,后面再正确也白瞎。我就不提汤剂和胶囊制备工艺上的巨大差别了,一句话,这基本就是两个不同的东西了。除非您能证明安慰剂效应比两种不同药物的差异性对结果影响还大,否则还是先正视问题,别忙着扯安慰剂的遮羞布了。
这样的临床有多大价值?非常低。
笑倒。这话放化药头上也合适,甚至更合适。化药可不是无色无味的哦,酸碱性刺激性特殊气味一应俱全,安慰剂的制作可大多只是用辅料简单代替药物就了事,比起中药安慰剂的制作,人家考虑得可少多了。
同样的话还给您:化药,薄膜衣片很多,但到了胃内还是会让患者感觉到,尤其是刺激性比较大的药物(不厚道的说,这种化药特别多)。这样患者就知道自己吃的是药物,还是安慰剂。
这样的临床试验有多大价值?大夫?
--------------------------------------------
这是我在本楼的最后一贴。再扯下去就成意气之争了。
临床试验不是我的专业方向,只是工作关系,有时涉及,但缺乏全面了解。我理解临床试验的必要性,相信双盲随机试验的科学性合理性。最开始提意见,是不认同您对中医评价指标的某些评论。中医评价指标一直有争议,我了解的也不多,看看您的观点,也觉得很有意思,启人思考。
不过,肠溶胶囊代替汤剂实在错到离谱。您花那么多工夫写文科普,这种错误还是少出为好。
降低人为差异,误差还要最重要的安慰剂效应。
给药途径和安慰剂效应那个更重要?安慰剂效应。
你从你的角度看给药途径困难。但是临床看最重要的如何不出现由安慰剂效应导致的bias。
不然为什么要做临床,用动物实验不就可以了。因为人有太明显的安慰剂效应。
不过,肠溶胶囊代替汤剂实在错到离谱。您花那么多工夫写文科普,这种错误还是少出为好。
看清楚重点是什么?安慰剂。
说用肠溶代替汤剂是你的说法。这是回避中药安慰剂的一个方法而已。下胃管也可以,肛门给药也可以,贴敷也可以。黑猫白猫,回避安慰剂效应就可以。
如果能够作出完全一致,色,形,味道,但是没有生理作用的安慰剂汤剂当然最好。
否则汤剂本身就完全受安慰剂效应影响。
临床不关心你用什么剂型,要得是患者不出现安慰剂效应导致的bias,也就是说知道,药物---导致疗效,而不是其他的东西。
临床实验需要控制的东西非常多,如果安慰剂效应都控制不了,这个临床实验,几乎没有价值。
测量是临床试验中比较关键的一个环节,谁来测量,测量几次,用什么方法,几个测量者,测量着是否是知道分组情况,跟踪的时间等等都会直接影响试验的质量。
通常最简单的方法是 两点测量,所谓的A-B线。A试验前的基础线 baseline, B试验后的变化。如果两组治疗组 同安慰剂组A1=A2,但是试验后A1〉A2就可以判定,药物有效。
但是实际试验中还有很多因素需要控制。
比如说测量数量,如果疾病有一定的自娱性就至少要测量三次,两次在试验前, A1,A2 间隔一段时间,然后是A3,如果A3=A2〉A1说明治疗没有意义,疾病的自愈性起到了决定性作用。比如说普通感冒,测量第一天,第七天,然后给药,测量第八天,(或者第十四天),第七天=第八,第十天〉第一天,就不能说药物有效。
再有如果疾病是慢性的,无法根治的就要长期跟踪,看看长期效果如何。典型的如糖尿病。
还是A1是基础线,A2 治疗后(比如血糖控制正常),A3 几个月之后,如果A1=A3《A2,就是能说,药物对疾病在短期内有效,长期看没有明显治疗价值。
燕轻笑提出的问题就是这个例子
我参加过一个皮肤病的新药的试验,一种是国产新药,一种是国外的药,不知道自己打的是哪种,打了以后三个月复查,效果很好。
结果一年以后我同一部位又复发了。
问:此复发记录是否对该药的检验结果有无影响?
在试验结论部分要写上,药物治疗观察三个月内效果如何,也就是说结果是有一定条件。
目前如果大家都是玩一个游戏,对疾病的预后不会出现太多的歧义。慢性病说三个月内有效,看的大夫就知道,这个试验是短期。因此在临床中就会有的放矢的使用。
测量人也非常有说道,几个人,水平如何,所谓inter, intra validity 如果试验报告中没有提到几个检测的人,说明试验设计不好,两个是最少,还要写上,之间的差异性。
测量的设备也很重要,不要说你用的就是好的,两种方式被介绍,1 大家公认的基标准,gold standard,比若说空腹血糖对糖尿病,NDI(neck disability index )对颈部疾患,(whaplash,颈椎病之类),MRI 对ACL 损伤,VISA 对髌腱病等。
因为这些都是经过反复试验检查的,业界公认。如果您说,用疼痛VAS来检查 whiplash,不是不可以,但是得出的结论就没有普遍意义,因为疼痛仅仅是NDI其中的一部分,还有很多的功能性指标,比如说提重物,睡觉等等。
另外就是MDC Minimal detective change 最小可测量的变化,就是说有效,无效是否被接受,不是测量者说了算,是其它的用这个测量方法的试验得出的结果。不允许老王卖瓜,自卖自夸。
这个看的比较清楚外链出处
PSFS patient specific functional scale 是临床用的非常多的针对患者功能的问卷,就是让患者写出来至少五个功能障碍,不论是疼痛,活动受限,还是压力,烦躁等等。
然后得出平均值,治疗后在做这个问卷,如果平均分提高2分,说明治疗有效,单个分数增加3分才有效。
如果得出每项都增加一分,看着很美,但是不被接受。
第二种方法是用新的标准,这个也可以,但是谁主张,谁举证。您可以用腋温来验证肩周炎的进展。但是要有足够的证据,证明腋温变化同肩周炎疗效有关。否则就不能被接受。验证一个新的检测方法也是非常难的,要同公认的金标准比较,要看 specificity 和 sensitivity。而且结果要能够被别人重现。
在这个领域不存在所谓的高手,你可以别人不行的。不能被重现就是不存在。可以参看康奈尔大学造假案和舍恩造价案。都是神奇不能被重复然后发现问题。
回到中医的话题,标准可以自己定,但是要研究同目前金标准的关系,不能说公说公有理,婆说婆有理。
另外要别人认可,重复的了。
现在看看所谓的验脉知男女,不论那一点都不能成为标准。当然脉诊,望诊都是一样的。
关于盲对于试验者的影响以前讲过了。最好是用外来的,第三方检测。实在不行,也要完全屏蔽任何知情的可能性,如果不是双盲结果至少没有双盲的有力。通常不能被采信。
注意这里是屏蔽检查者知情,不是治疗者。针灸师可以知道治疗手段,但是不一定是检查者。
最后介绍所罗门四分组试验:
该设计也是只有一种实验处理,随机选择被试和分组;一共4个组,两个实验组,两个控制组;两个实验组中,一个组有前测与后测,一个组只有后测;两个控制组中,也是一个组有前测与后测,另一个组只有后测。在上面的基本模式中第1、2 组是实验组,第3、4组为控制组;第1、3组有前测与后测,第2、4组只有后测。
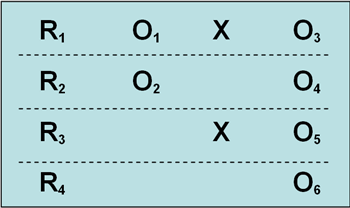
这个试验的好处是最大程度上回避了检验着对结果的主观影响。
缺点是分组太多试验费用,周期增加。
总之,看一个试验报告,并不是简简单单看结果,要看很多细节。
这也就是为什么要做SR,作Cochrane reviews因为它们有更多时间,经验关注细节,联系作者,是普通临床医生无法比拟的。也就是说他们是RCT的检查者。
普通医生没有时间,精力去认真检查每一个报告,因为看Cochrane reviews 是临床医生公认的简单,快捷的评估相关领域,目前最有效证据的方法。
这也是,为什么EBM证据标准把cochran reiews 放到第一位的原因。



想批驳cochrane reviews 最好先把,最基础的概念搞清楚在出来说话。
本帖一共被 1 帖 引用 (帖内工具实现)
比如监管部门允许发一个新药或者一种新的诊疗方式,是必须有最高级的证据呢,还是次一级的也可以接受?
各国药监都要求做RCT才给新药审批的。不存在次一级的问题。
FDA:
The trials are typically conducted in three phases:
* Phase 1: The drug is tested in a few healthy volunteers to determine if it is acutely toxic.
* Phase 2: Various doses of the drug are tried to determine how much to give to patients.
* Phase 3: The drug is typically tested in Double-blind, placebo controlled trials to demonstrate that it works. Sponsors typically confer with FDA prior to starting these trials to determine what data is needed, since these trials often involve hundreds of patients and are very expensive.
* (Phase 4): These are post-approval trials that are sometimes a condition attached by the FDA to the approval.
The legal requirements for safety and efficacy have been interpreted as requiring scientific evidence that the benefits of a drug outweigh the risks and that adequate instructions exist for use, since many drugs are toxic and technically not "safe" in the usual sense.
中国SDA
任何的单独病例报道式的,都绝对不可能得到审批的。
我们这里谈的是从临床角度上看。大多数都是已经上市的药物,进行比较,为啥用A不用B。
这就要看证据的级别了。
这也是为什么要做EBM的原因,说服太多,总要有一个标准才好,否则公说公有理,婆说婆有理。到底听谁的。
会EBM的临床医生要至少有两种能力。
1:会critique article, 自己独立能够评估医学文献。
但是这个还有很多的局限性,比如说时间,对文献的资料掌握,很多的文献是要钱的,对文献深度的掌握,您不能每个都联系员是作者,而且人家也未必打理你。
2:要会看证据的级别。不是说cochrane reviews就是圣经,而是人家资料全,深度高,经验丰富,得出的结论比自己做的可靠性好。
但是反过来,如果没有SR,没有cochrane reivews就要自己做,如果SR或者cochrane reviews 的内容同您的患者人群不一致,也需要自己做,比如说,您要给小孩子看,但reviews的是大人小孩都有,或者仅仅是大人,就没有办法作为证据。
但是不论如何,会EBM的临床医生到要至少会评估文献,要自己会写论文。这样才知道文献说的是什么。
至于病例报道,专家意见(不是所谓的专家,是在该研究领域的专家,尤其是临床研究的),患者选择都要低于SR更低于cochrane reviews.
以后有机会还会讲到如何评估SR。
这些都有评分标准的。
给人血液中注射牛奶、酱油之类的液体,会带来什么不良后果?
任何异体蛋白进入血液都会激活免疫反应。严重的会死人。
如果少量的几个ml会有反应,但未必会死人。
往血管里注射东西不是闹着玩的。